Genome level calculate Ka/Ks --gKaKs
The main purpose of this program is to align the CDSs from a well-annotated genome to a target genome, which hasn’t been annotated yet. It uses blat to find the best match between the CDSs and target genome sequences, and then uses bl2seq to align every CDS to the blat-identified target genome region. After merging the aligned sequences and removing gaps according to reference CDS codon, it uses codeml/yn00 of PAML and other 10 methods from KaKs_Calculator to compute the ka/ks ratio between CDSs and their homolog sequences in the target genome. Also this program can compute Ka/Ks ratio for two lists of homologous DNA sequences, with one list as CDS sequences and the other list as genomic sequences.
Program installation:
This program require several widely used bioinformatics programs. Before running this program, please make sure the installation of the bellowing programs. We suggest to make a link at /usr/local/bin/.
Data preparation:
- The CDS sequences of the reference genome in fasta format –query_seq=”ref.cds”
- The gff3 file of the reference genome –gff=”ref.gff3”
- Target genome sequence in fasta format –hit_seq=”hit.fa”
Flowchart:
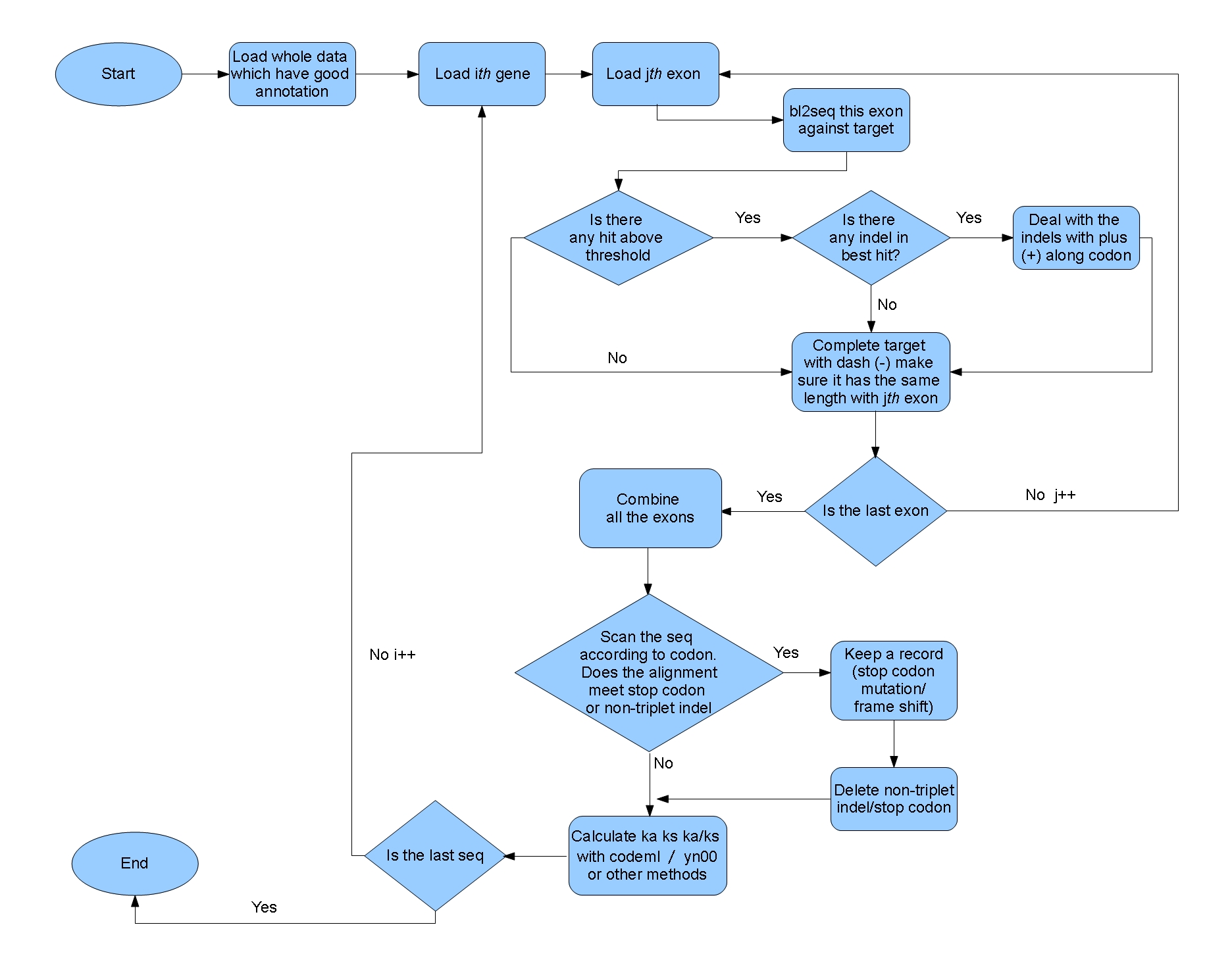
The function of the pipeline:
This program can be used to compute ka/ks ratio between the genes in one well-annotated genome and their ortholog sequences in another closely related genome, which hasn’t been annotated. The result
- can be used to compute the diverge time between two species through estimating average Ks and mutation rate;
- can be used to estimate how many ortholog sequence pairs are under functional constraints;
- can be used as evidence to annotate genes
Notation:
- This program works for closely related species. If species splits from each other too long ago, the sequences diverge strikingly, bl2seq cannot generate good results.
- Because this program deletes all the gaps that can’t be aligned in codon level, the Ks generated by this approach tend to be smaller.
- In –problem_loc records, the Ks/Ka cannot be computed for the CDS id under ===reverse=== category.
- More details please read document.